Mukopolisacharydoza

Mukopolisacharydoza - grupa uwarunkowanych genetycznie chorób wynikających z zaburzeń metabolicznych kwaśnych mukopolisacharydów (glikozaminoglikanów). Dziedziczenie jest autosomalne recesywne sposób. Określony przez chorobę układową szkieletu i opóźniony rozwój fizyczny. W niektórych formach Mukopolisacharydozę obserwowano opóźnienie umysłowe. Ewentualne wady serca, patologii widzenia, tworzenie przepuklin, zaburzeń neurologicznych, nadmierne owłosienie, powiększenie wątroby i śledziony. Rozpoznanie opiera się na charakterystycznych objawów klinicznych, radiografii i innych badaniach danych. Leczenie jest objawowe. Rokowanie jest słabe, tak jak we wszystkich formach mukopolisacharydozy obserwowano postęp zmian patologicznych w wyniku ciągłej kumulacji glikozaminoglikanami.
Mukopolisacharydoza
Mukopolisacharydozy - grupę chorób genetycznych związanych z akumulacji mukopolisacharydów kwasu w narządach i tkankach. Przyczyną rozwoju jest wrodzonym niedoborem enzymów lizosomalnych. Po raz pierwszy Mukopolisacharydoza Hurlera został opisany w 1917 roku. Mukopolisacharydoza leczenie ortopedyczne odbywa się z udziałem kardiologów, okulistów, neurologów, Otolaryngolodzy i innych specjalistów.
Typ Mukopolisacharydoza IH
Typ Mukopolisacharydoza IH lub zespół hurler występuje u 1 noworodka od 20-25 tys. Objawy Mukopolisacharydoza pojawić w pierwszym roku życia, pełny obraz kliniczny jest utworzony do 1-2 lat. Dla danej postaci charakteryzuje Mukopolisacharydoza grube cechy twarzy i odkształcenie czaszki w postaci kuter (skarotsefaliya). Ze względu na zwiększone adenoidy i wady rozwojowe jamy ustnej pacjentów twarz i nos oddychaniem. Odnotowano postępy deformacje kończyn i innych części układu kostnego, karłowatości.
Kręgosłup pacjentów z Mukopolisacharydozę zakrzywiona, z powodu tego, co pojawia się objaw pozycji siedzącej z tyłu „kota”. Wyraźne skrócenie szyjki, wysokie położenie ostrza oraz vystoyanie dolne żebra. szerokiego pędzla jak pazur. Z biegiem czasu, pacjenci z Mukopolisacharydozę utworzona przykurcze stawów. początkowo dotknięte łokieć i stawów ramię, a następnie - kolano, biodrowego i stawu skokowego. Ze względu na ograniczenia ruchów jest charakterystyczny chód - na palcach, ze zgiętymi nogami.
Tony serca są przytłumione, granice rozszerzony. Osłuchiwanie określona skurczowe hałas na EKG wykazała rozproszone uszkodzenia mięśnia sercowego. Przedniej ściany jamy brzusznej pacjentów z mukopolisacharydozy osłabiony żołądka zwiększona, często wykrywane wodniak, pępkowy i przepuklina pachwinowa. Wątroby i śledziony w powiększeniu. Charakteryzuje się zaburzeniami wzroku i słuchu. Możliwe zmętnienie rogówki i wzrostu, z pigmentem dystrofia siatkówki, jaskra, zanik nerwu wzrokowego i stagnacji w obszarze dna. U chorych na Mukopolisacharydozę typ to się postępującym upośledzeniem umysłowym. Nie może być upośledzona koordynacja ruchów, niedowłady i porażenia. Wykazała nadmiernego wzrostu włosy mieszkowe włosów.
kręgosłupa rentgenowskie pacjentów z MPS przedstawia charakterystykę deformacji kręgosłupa. Kręgi mają kształt prostopadłościanu, ich kontury są zaokrąglone w sekcji przejściowej jest wykryta zbieżność przedniej kąty angulate kifoza, pogrubienie i skracania pędów. Na RTG klatki piersiowej jest określana przez pogrubienie przednich i tylnych części przerzedzenie krawędziach, towarzyszyć ich łyżeczkowate lub odkształcenia szabli, skróceniu i deformacji obojczyków i zmniejszyć przemieszczenie głowic kości ramiennej.
Radiografia potwierdza miednicy skurcz redukcji pierścienia miednicy, panewki stożkowe i głowice udowej. Radiologiczne kości długich ujawnił korowej przerzedzenie i ekspansję jamy szpikowej. RTG kości ręki pokazuje niedorozwój falangi paznokci, skrócenie i rozbudowa kości śródręcza, proksymalnej i środkowej paliczków. Na zdjęcia rentgenowskie czaszki twarzowej niedorozwój kości, zdefiniowanego craniostenosis i macrocephaly.
Mukopolisacharydoza typ I-S
Mukopolisacharydoza-S typu I lub chorobę Sheye (opisany w 1962 roku przez American okulisty Sheye) jest nowsza, względnie korzystne przepływające jeden rodzaj mukopolisacharydozy H (zespół Hurler). rozwój 3-6 lat dzieci jest prawidłowe. Pierwszą oznaką Mukopolisacharydozę stać przykurcze zgięcie palców. Następnie ograniczone rozszerzenie w nadgarstku, łokieć i ramię stawów. Przykurcz kończyn dolnych są zwykle łagodne. Pełny obraz kliniczny Mukopolisacharydozę tworzą początek dojrzewania.
Pacjenci z Mukopolisacharydozę krępy, niski, grube rysy twarzy i dobrze umięśnione. Istnieje zwiększone owłosienie (owłosienie). Często nie są pachwinowej lub przepukliny pępowinowej. Skóra na palcach rozciągnięte i zgęstnieje. Ze względu na kompresję nerwu może rozwinąć zespół cieśni nadgarstka, towarzyszy zanik mięśni kłębu kciuka regionu i parestezje w obrębie palców III-IV. Niektórzy pacjenci z Mukopolisacharydozę ujawnione zwężenie zastawki aortalnej, niewydolność zastawki aortalnej, pigmentowe zwyrodnienie siatkówki, jaskra i zmętnienie rogówki. Inteligencja jest na prawidłowym, powiększonym śledziony i wątroby nietypowo. Radiologicznie zdefiniowany wzorzec podobny typ Mukopolisacharydoza IH, jednak zmiany patologiczne są mniej ostre.
Mukopolisacharydoza typu II
typ II lub Mukopolisacharydoza zespół Hunter najczęściej diagnozowane u chłopców i zazwyczaj rozwija się w życiu 2-3 lat. Jak z Mukopolisacharydozę typu IH obserwowane scaphocephaly, wulgaryzowaniem rysów twarzy, głębokim głosem i trudności z oddychaniem z powodu deformacji twarzoczaszki. Kiedy nie jest to cecha typu Mukopolisacharydoza H kifoza typowo wykrywa objaw „CAT powrotem” negatywne. Pacjenci często cierpią na zakażenia dróg oddechowych (zapalenie oskrzeli, tchawicy, zapalenie płuc). Stopniowo rozwijać zaburzenia koordynacji ruchowej, agresja wzrasta. Niestabilny nastrój, z szybkimi zmianami.
Także w tej postaci mukopolisacharydozy istnieje niewielkie powiększenie wątroby i śledziony, postępujące utratę słuchu, guzki na skórze pleców i spadkiem inteligencji. Następnie, może wystąpić zmętnienie rogówki. zdjęcie rentgenowskie - jak w Mukopolisacharydozę typu I-S. Istnieją dwa warianty tej choroby: korzystne (wariant B) i korzystne (wariant A). Z korzystnych wariantów objawy są łagodne, czasem istnieje niewielkie opóźnienie umysłowe, z Mukopolisacharydoza pacjenci mogą żyć do 30 lat lub więcej. Dla najgorszym przypadku charakteryzuje się jasnym obrazem klinicznym i poważnych naruszeń inteligencji. Zabójcza wynik pojawia się w okresie dojrzewania.
Mukopolisacharydoza Type III
Mukopolisacharydoza typu III lub chorobą Sanfilippo (opisany w 1963 roku przez amerykańskiego pediatrę Sanfilippo) występuje u noworodka 1 100-200 tys. We wczesnych latach rozwoju życia odpowiedni do wieku, są niekiedy trudności w połykaniu i niezgrabny chód. W wieku 3-5 lat dziecko z MPS staje się apatyczny i zaczyna pozostawać w tyle w rozwoju. Są to zaburzenia mowy, coarsening rysów twarzy, nietrzymanie moczu i kał. Z biegiem czasu, postępujące upośledzenie umysłowe i zajmuje centralną pozycję w obrazie klinicznym tego typu Mukopolisacharydozę.
Wraz z zaburzeniami umysłu zaobserwowano umiarkowane powiększenie wątroby i śledziony, zwiększony wzrost włosów, skurcz i zahamowanie wzrostu. Patologia oczu i układu krążenia dla tej formy Mukopolisacharydozę nietypowym. zdjęcie rentgenowskie - bez zmian lub co Mukopolisacharydoza typ I-S, ale mniej wyraźne. Pacjenci z Mukopolisacharydoza trzeciego typu zwykle umierają w wieku od 10-20 lat zakażenia.
Typ Mukopolisacharydoza IV
Typ Mukopolisacharydoza IV lub choroba Morquio (opisany w 1929 roku przez Urugwajskiego pediatry Morquio) występuje u 1 na 40000 noworodków. Do 1-3 lat, dzieci rozwijają się normalnie. Następnie, istnieje znaczne opóźnienie wzrostu, skrócenie szyi i tułowia i przykurczów koślawe zniekształcenie kończyn, skolioza lub kifozę, różnorodne deformacja klatki piersiowej, zmniejszenie siły mięśniowej, zgrubienie skóry i brutalizacji rysów twarzy. Z tego typu Mukopolisacharydozę często rozwijają pachwinowy i przepukliny pępowinowej, dystrofię rogówki i utraty słuchu. Inteligencja jest przechowywany.
Na rentgenowskie kręgosłupa określa kifozy, skolioza, rozszerzając i spłaszczenie trzonów kręgów. W prowadzeniu rentgenowskie miednicy i kończyn wykazały wielokrotne odkształcenie, nierównych konturów spłaszczenie głowy kości udowej, skrócenie kości przedramienia i deformacje stóp. Średnia długość życia chorych na Mukopolisacharydozę - mniej niż 20 lat. Śmierć w tej formie Mukopolisacharydozę występuje z powodu chorób współistniejących komplikuje niewydolności sercowo-płucnej.
Inne rodzaje Mukopolisacharydozę
Mukopolisacharydoza typu VI lub choroba Maroto-Lamy (opisany w 1960 roku przez francuskiego Lamy i Maroto) rozwija się w wieku 2 lat i starszych. Jest coarsening rysów twarzy, opóźnienie wzrostu, skrócenie szyjki, przykurcze stawów i deformacji klatki piersiowej baryłkę. częstymi przeziębienie. Możliwe przepukliny, powiększenie wątroby i śledziony. Inteligencja nie jest naruszona. Istnieją dwie opcje przepływu: klasyczny i Mukopolisacharydoza z łagodnymi objawami klinicznymi. Na zdjęcia rentgenowskie pacjentów z Mukopolisacharydozę określona prostopadłościan lub Klinowy kształt deformacji kręgosłupa, trójkątny miednicy deformacja, niedorozwój i deformacja głowy kości udowej, strzałkowy skracanie.
Mukopolisacharydoza typu VII Postępowano jak mupokolisaharidoz typu III występują różnice tylko w badaniach biochemicznych. Typ Mukopolisacharydoza VIII objawowo przypomina Mukopolisacharydoza typu IV, lecz, przeciwnie, towarzyszy niedorozwój umysłowy.
Diagnostyka i leczenie Mukopolisacharydozę
zestaw diagnoza Mukopolisacharydoza na podstawie charakterystycznego obrazu klinicznego i radiologicznego, wykrywanie glikozaminoglikanów w moczu i aktywność enzymu badania w hodowlach komórkowych. W badaniu pacjentów z Mukopolisacharydoza powołanych różnych ekspertów konsultowany: .. kardiolog, gastroenterologa, okulista, laryngolog, neurolog, psychiatra, itd., Są przeprowadzane badania instrumentalne, aby ocenić stan poszczególnych narządów i układów.
Terapia patogenetyczne Mukopolisacharydoza być rozwijane. Leczenie jest objawowe, może to być zarówno zachowawcze i operacyjne. Zapobieganie i zarządzanie infekcje dróg oddechowych, korekcja utraty wzroku i słuchu. W razie potrzeby i przepuklina naprawy wykonywane operacje hernioplasty wyeliminować przykurcze i korekcję deformacji kostnych. Rokowanie wszystkich rodzajów Mukopolisacharydozę niekorzystnych - kontynuacja akumulacja metabolitów w tkankach prowadzi do nasilenia patologicznych zmian ze strony wszystkich narządów i układów. Stosowanie jakichkolwiek środków terapeutycznych (transfuzji krwi, podawanie hormonów itd. D.) z Mukopolisacharydozę zapewnia tylko chwilowy poprawę. Zaleca opieki prenatalnej.
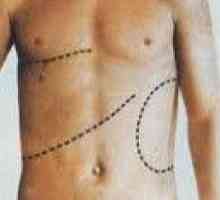 Powiększenie wątroby i śledziony
Powiększenie wątroby i śledziony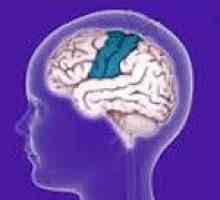 Łagodny padaczka rolandic
Łagodny padaczka rolandic Miopatia wrodzona
Miopatia wrodzona Chorobę Machado-Josepha
Chorobę Machado-Josepha Platibaziya
Platibaziya Zespół Sturge-Webera
Zespół Sturge-Webera Kardiomiopatia wtórna
Kardiomiopatia wtórna Zespół Hunter
Zespół Hunter Fenyloketonuria
Fenyloketonuria Kolagenazy
Kolagenazy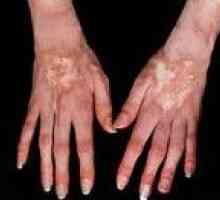 Twardzina układowa
Twardzina układowa Utrata pola widzenia
Utrata pola widzenia- Nadczynność przytarczyc
 Talasemia
Talasemia Niedokrwistość hemolityczna
Niedokrwistość hemolityczna- Terapia komórkowa
- Zespół Turnera
 Diament zespół shvahmana
Diament zespół shvahmana- Niezaspokojone pragnienie stałej
 Elektroretinografia
Elektroretinografia- Wrodzone wady i chromosomalnych nieprawidłowości płodu
 Platibaziya
Platibaziya Groźne zakażenia w czasie ciąży
Groźne zakażenia w czasie ciąży Niedokrwistość hemolityczna
Niedokrwistość hemolityczna Zespół Sturge-Webera
Zespół Sturge-Webera Powiększenie wątroby i śledziony
Powiększenie wątroby i śledziony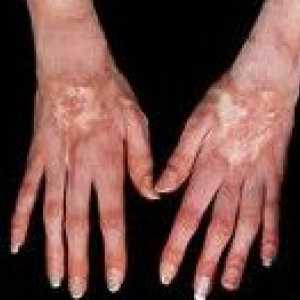 Twardzina układowa
Twardzina układowa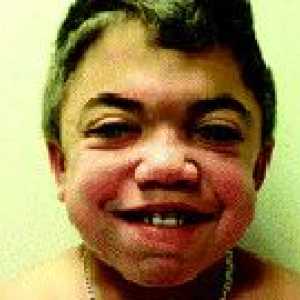 Zespół Hunter
Zespół Hunter