Choroba von Hippel Lindau

Choroba von Hippel Lindau - autosomalny dominujący nieprawidłowości genetycznych powodując rozwój w organizmie szereg nowotworów polimorficznych. Najczęściej naczyniak siatkówki hemangioblastoma OUN, guza chromochłonnego, nowotwory nerki i raka trzustki. Czasami objawem choroby działa jako pojedynczy proces nowotworu. Diagnozę zweryfikowano po neurologicznych i okulistycznych, badanie CT lub MRI mózgu i kręgosłupa, ultradźwięków lub CT nerki, trzustki, nadnerczy, diagnostyki genetycznej. Leczenie polega na wczesne wykrywanie i usuwanie powstającego powstawania nowotworu.
Choroba von Hippel Lindau

Choroba występuje z częstością 1 na 36 tys. Ludzi. Określony większa polimorfizmu różnej lokalizacji nowotworów powstać. Najczęstszym objawem jest retinal angiomatosis który towarzyszy aż do 75% przypadków. Często pojawia się marker diagnostyczny dla tej choroby. naczyniak krwionośny zarodkowy Móżdżek według różnych źródeł obserwuje się w 35-70% przypadków nowotworów i torbieli nerkowych - 25% pacjentów, pokonaniu trzustki - 24% pheochromocytoma - 7%. Ze względu na dużą różnorodność nowotworów u pacjentów z chorobą von Hippel-Lindau, wymaga wspólnej kontroli ekspertom w dziedzinie okulistyki, onkologia, neurologia, urologii, gastroenterologii, endokrynologii.
Przyczyny tej choroby von Hippel-Lindau
choroba Von Hippel Lindau jest chorobą genetyczną. Około 80% z nich jest dziedziczona w sposób autosomalny dominujący sposób z niepełnego genu penetracji. Kolejne 20% choroby von Hippel-Lindau występuje z powodu nowych mutacji. Aberracje koncernu znajduje się w chromosomie r25-26 3 części, a mianowicie VHL gen, który odgrywa rolę tłumika, który hamuje wzrost guzów. Do chwili obecnej istnieje około 140 mutacji tego genu.
W rezultacie, nie ma wystarczającej onkosupressii wzrost nowotworowy, korzystnie angioretikulom naczyniak krwionośny zarodkowy. Guzy wpłynąć móżdżku i siatkówki oka, rzadko obserwowane Śródmózgowy nowotworowe półkule mózgu, nowotwory podkorowych struktur i rdzenia przedłużonego, jeszcze rzadsze - guz rdzenia kręgowego i nerwów obwodowych. Ze względu na niepełną przejaw aberracji genetycznych jedynym klinicznym objawem choroby można zaobserwować u niektórych pacjentów.
Zgodnie z klasyfikacją, choroba von Hippela-Lindaua jest typu 2 guza chromochłonnego i bez jego obecności. Drugi typ jest podzielony na wersje: 2a - przy niskim ryzyku gruczolakoraka nerek, 2B - rak wysokiego ryzyka, 2C - istnieje tylko guz chromochłonny. We wszystkich przykładach, z wyjątkiem choroby 2C, może naczyniak krwionośny zarodkowy OUN i siatkówki naczyniaki.
Objawy choroby Von Hippel-Lindau
Objawy neurologiczne debiut zwykle spada na 3-4th dekadach życia. W dzieciństwie, choroba von Hippel-Lindau charakteryzuje się występowaniem objawów neurologicznych na tle już istniejących zaburzeń wizualnych. W niektórych przypadkach choroba objawia się u dzieci krwotok podpajęczynówkowy.
CNS. Najczęstsze objawy stanowią główne źródło móżdżku cyst (torbieli móżdżku). One przejawiać objawy mózgowe (rozlane bóle głowy, nudności, bez względu na przyjmowanie pokarmu, wymioty, szumy uszne), spowodowane przez wzrost ciśnienia śródczaszkowego. Były również wyróżniona epipristupy, mogą być uogólnione lub ogniskowej. Z biegiem czasu, objawy móżdżku formowanie objawu ataksja móżdżkowa: Statyczne i dynamiczne discoordination, adiadohokinez, hypermetric i hyposynergia zamiar drżenie, miodistoniya. Jak wzrost nowotworów występuje przemieszczenie móżdżku i kompresji z pnia mózgu, towarzyszą objawy macierzyste, głównie zaburzenia połykania, podwójne widzenie, dyzartria. guzy rdzenia (najbardziej angioretikulomy) pojawiają osiowe zespoły, utrata głębokiej wrażliwości gatunku, brak odruchów ścięgnistych. W 80% przypadków patologii kręgosłupa notatek kliniki, podobny do Syringomielia. Możliwe obraz kompletnej klęsce przekroju rdzenia kręgowego.
choroby oczu zdiagnozowana we wczesnym stadium tylko w oftalmoskopii. Po 8 latach, są skargi dotyczące obrazu Nebula i jego zniekształceń (metamorphopsia). Połowa pacjentów wykazały zarówno choroby oczu. Zwiększenie siatkówki naczyniak ostatecznie doprowadzić do załamania się krążenie krwi w jej naczyniach krwionośnych, niedokrwienia i torbielowate zwyrodnienie. W późniejszym etapie ewentualnej zapalenie błony naczyniowej oka, zaćma, retinal disinsertion, jaskra, hemophthalmus.
uszkodzenie nerek w 60-90% przypadków przedstawione torbiele w 45% przypadków - rak renalnokletochnoy. Zwykle, rak nerki klinicznie debiut w wieku 40 do 50 lat u pacjentów uprzednio leczonych z powodu nowotworów. W połowie przypadków w momencie rozpoznania raka wykazały przerzuty. kombinacja torbielowatość nerek z siatkówki angiomatosis jest bardziej powszechne niż jego połączeniu z naczyniaków mózgowych. U 35% pacjentów z chorobą von Hippel-Lindau, zespół policystycznych zdiagnozowana pośmiertnie. U dzieci w rodzinnej typu choroby policystycznych nerek jest często jedynym objawem jest.
pheochromocytoma prawie połowa przypadków ma charakter dwustronny. Może on działać tylko klinicznej manifestacji choroby. W połączeniu z rakiem nerki występuje rzadko.
trzustka porażka od 30% do 72 stanowią torbieli. Torbiele trzustki Są łagodne i rzadko powodują klinicznie istotnego niedoboru enzymów trzustkowych. Choć zdarzają się przypadki całkowite zastąpienie tkanki normalnego rozwoju torbieli gruczołu krokowego cukrzyca.
Rozpoznanie choroby von Hippel-Lindau
Pełna weryfikacja diagnozy wykonanej wspólnie przez neurologa, okulisty, genetyk z udziałem innych lekarzy: onkologa, urologa, endokrynologa, gastroenterologa.
Na początkowym etapie pełnego neurologicznych i badania okulistycznego. W celu wykrycia móżdżku formacje powołać CT lub MRI mózgu. Do wykrywania nowotworów w innych miejscach należy ultradźwięków lub CT nerek, trzustki USG lub rezonans magnetyczny, MRI kręgosłupa, nadnerczy, CT. Analiza poziomu katecholamin i enzymów trzustkowych. Rozpoznanie DNA ma na celu identyfikację mutacji w VHL genu.
Zakładamy i wykluczyć chorobę von Hippel-Lindau powinna w każdym przypadku angiomatosis siatkówki podczas oftalmoskopii, zwłaszcza gdy istnieje historia rodziny. W początkowym etapie oftalmoskopii można określić za pomocą jednego angiomu rozszerzenie naczyń siatkówki wprowadzenie go następnie się wiele naczyniaki, znamienny kręte krętość tętniaka naczyniowego. Diagnozowanie najwcześniejsze zmiany w naczyniach siatkówki i forma skasowane pozwala angiografii fluorescencyjnej siatkówki. Może być stosowany do różnicowania siatkówki zmiany towarzyszące chorobie von Hippel Lindau od innych schorzeń okulistycznych: retinopatia, ritinita, siatkówczaka, neuropatia nerwu wzrokowego i tak dalej. precyzyjna diagnoza jest możliwe przy użyciu tomografii laserowej siatkówki.
Leczenie i rokowanie choroby Von Hippel-Lindau
Obecnie choroba von Hippel-Lindau jest tylko leczenie objawowe. Ma to na celu wyeliminowanie guza wystąpić formacji. Podobnie jak w przypadku rocznej skriningu i obserwacji pacjentów może być bardziej wczesnego wykrywania nowotworów.
Wczesne stadia siatkówki angiomatosis jest wskazaniem do radioterapii ogniskowej, ale rok po jego realizacji może być promieniowanie retinopatia. Ze względu na małe rozmiary możliwe naczyniaki fotokoagulacji laserowej, diatermii, z szerszej edukacji - transscleral cryopexy. Jeśli choroba von Hippel-Lindau towarzyszy nowotworów OUN, należy skonsultować się z neurochirurga.
Możliwe chirurgiczne usunięcie guz móżdżku, półkule mózgu, nerwu wzrokowego. Przypadki stosowania chirurgii stereotaktycznej. Podczas diagnozowania raka nerek przeprowadzono nefrektomię częściową, do wykrywania guza chromochłonnego - jego usunięcia. Leczenie łagodnych nowotworów trzustki pokazano poprzez zwiększenie wielkości powyżej 2-3 cm.
Bez leczenia postęp choroby prowadzi do utraty wzroku z powodu stopniowego siatkówki angiomatosis i krytyczny ze względu na rozwój nowotworów mózgu i lokalizacji somatycznej. W obserwacji i leczenia pacjentów żyją średnio 40-50 lat. Połowa zgonów spowodowanych przez centralny nerwowego hemangioblastoma systemu. We wczesnych stadiach rodnika usunięcie nowotworu jest możliwe w większości pacjentów, jednak, guzy mają tendencję do nawrotów po około 6 lat od ich usunięcia.
 Trzustka wzrosła echogeniczności. Co to znaczy i jak to leczyć?
Trzustka wzrosła echogeniczności. Co to znaczy i jak to leczyć? Rak trzustki
Rak trzustki Rak jelita
Rak jelita Liści nowotworu sutka
Liści nowotworu sutka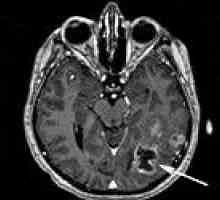 Guzy ośrodkowego układu nerwowego
Guzy ośrodkowego układu nerwowego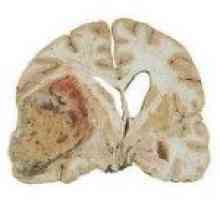 Wewnątrzmózgowe guzy półkul mózgowych
Wewnątrzmózgowe guzy półkul mózgowych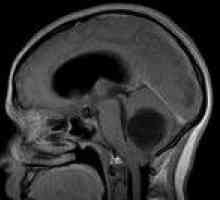 Hemangioblastoma
Hemangioblastoma Zespół Louis-Bar
Zespół Louis-Bar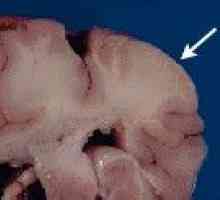 Fakomatozą
Fakomatozą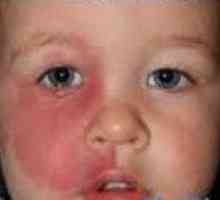 Zespół Sturge-Webera
Zespół Sturge-Webera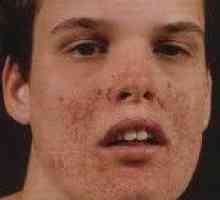 Stwardnienie guzowate
Stwardnienie guzowate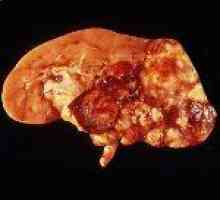 Rak clear-cell
Rak clear-cell Rak clear-cell
Rak clear-cell Rurowy rak nerki
Rurowy rak nerki Rdzeniasty rak tarczycy
Rdzeniasty rak tarczycy Pierwotne nowotwory wielokrotne
Pierwotne nowotwory wielokrotne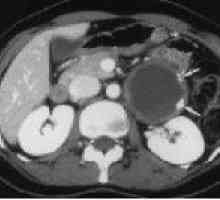 Vneorgannye zaotrzewnowy guza
Vneorgannye zaotrzewnowy guza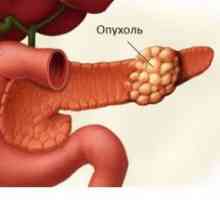 Rak trzustki
Rak trzustki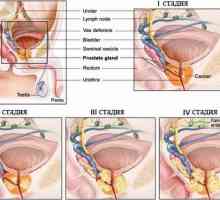 Rak prostaty
Rak prostaty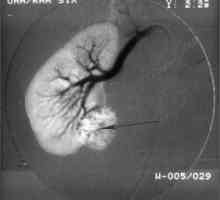 Rak clear-cell
Rak clear-cell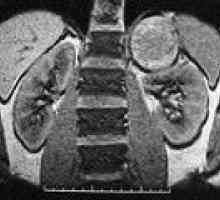 Pheochromocytoma
Pheochromocytoma
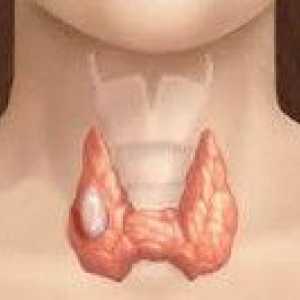 Rdzeniasty rak tarczycy
Rdzeniasty rak tarczycy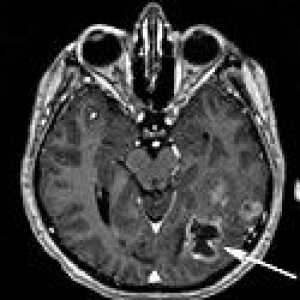 Guzy ośrodkowego układu nerwowego
Guzy ośrodkowego układu nerwowego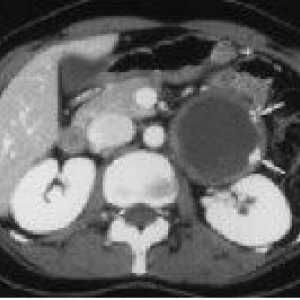 Vneorgannye zaotrzewnowy guza
Vneorgannye zaotrzewnowy guza Pierwotne nowotwory wielokrotne
Pierwotne nowotwory wielokrotne Zespół Louis-Bar
Zespół Louis-Bar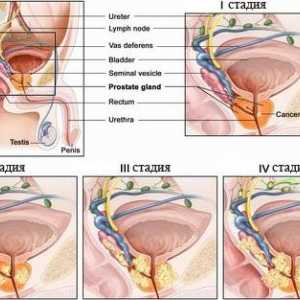 Rak prostaty
Rak prostaty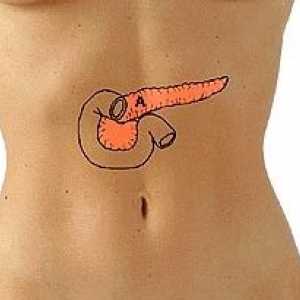 Więcej niż 4% komórek trzustkowych nie dają walce z rakiem
Więcej niż 4% komórek trzustkowych nie dają walce z rakiem Zespół Sturge-Webera
Zespół Sturge-Webera Pheochromocytoma
Pheochromocytoma Rurowy rak nerki
Rurowy rak nerki