Autoimmunologiczny zespół wielogruczołowy

Autoimmunologiczny zespół wielogruczołowy - endokrynopatią autoimmunologicznym występujące przy jednoczesnym pierwotnym uszkodzeniem wielu gruczołów dokrewnych i innych narządów. Autoimmunologiczne zespół wielogruczołowy typu 1 oznaczony niewydolność nadnerczy, kandydozę z skóry i błon śluzowych, gipoparatireoz- autoimmunologicznych wielogruczołowy typu zespół 2 wpływy z rozwojem niewydolności nadnerczy, hiper- lub tarczycy, cukrzyca insulinozależna, pierwotnej niedoczynności gonad, miastenia gravis, steatorrhea wsp. Naruszeń. Diagnoza obejmuje określenie zestawu parametrów laboratoryjnych (biochemia krwi i hormonów w moczu), ultradźwiękami nadnerczy i CT, USG tarczycy. Leczenie autoimmunologicznego zespołu wielogruczołowego wymaga wyznaczenia hormonalnej terapii zastępczej (kortykosteroidów i mineralokortykoidy, L-tyroksyny).
Autoimmunologiczny zespół wielogruczołowy

Autoimmunologiczne wielogruczołowy zespół (APG) - zaburzenia immunoendokrinnoe charakteryzuje podstawowej czynnościowe kilku gruczołów dokrewnych, nie dokrewnymi chorób narządów specyficzne. W endokrynologii odróżnić autoimmunologiczny zespół wielogruczołowy typu 1 i 2 (związki APG-y 1, 2), które mają genetyczne, immunologiczne i kliniczne cechy.
Autoimmunologiczne zespół wielogruczołowy typu 1 zazwyczaj objawia się w dzieciństwie (10-12 lat), a czasami określany w literaturze także terminem „młodzieńczego rodziny poliendokrinopatiya”. Całkowita częstość występowania APG-1 jest mały, ale choroba jest nieco bardziej powszechne w populacji mężczyzn, głównie mieszkańców Finlandii, Sardynia, Iran, z powodu długotrwałej izolacji genetycznej tych ludów. Autoimmunologiczne zespół wielogruczołowy typu 1, składa się z triady objawów: niewydolność nadnerczy, gipoparatiroz i kandydoza.
Najczęstszym wariantem wielu autoimmunologicznych endokrynopatii jest autoimmunologiczną zespół wielogruczołowy typu 2, która rozwija się u osób dorosłych (w wieku 20-30 lata) - Kobiety dominują wśród pacjentów. APG są elementy 2 niewydolność nadnerczy, cukrzyca Typu 1, autoimmunologiczne zapalenie tarczycy zaangażowanie według rodzaju pierwotnej niedoczynności lub nadczynności tarczycy.
Oprócz zaburzeń endokrynologicznych, autoimmunologiczne zespół wielogruczołowy typu 1 i 2 w połączeniu z innymi przejawami narządowo-specyficzne.
Przyczyny autoimmunologicznym zespołem wielogruczołowy
Oba typy autoimmunologicznego zespołu wielogruczołowego są uwarunkowane genetycznie, jak wskazano przez rodzinę oraz chorób dziedzicznych. Zatem APG-1 wpływa braćmi i siostrami jednej generation- autoimmunologiczny zespół wielogruczołowy typu 2 - przedstawicieli tej samej rodziny od kilku pokoleń.
Autoimmunologiczne zespół wielogruczołowy typu 1 - jedynym znanym choroby autoimmunologiczne o monogenowe w przyrodzie. Rozwijać APG-1 mutacji genu prowadzi autoimmunologiczne regulatora (AIRE), znajduje się na długim ramieniu chromosomu 21 (21q22.3). Autoimmunologiczne wielogruczołowy zespół 1 jest dziedziczona w sposób autosomalny recesywny sposób i nie jest związane z haplotypów HLA.
Autoimmunologiczne zespół wielogruczołowy typu 2, wiąże się z HLA haplotypu (antygeny, DR3, DR4, DR5, B8, DW3). Jest prawdopodobne, że mechanizm rozwoju APG-2 wiąże się z nieprawidłową ekspresją antygenów HLA systemu na błonach komórek gruczołów wydzielania wewnętrznego, która działa pod wpływem wszelkich czynników zewnętrznych.
Objawy autoimmunologiczne zespół wielogruczołowy typu 1
Autoimmunologiczne zespół wielogruczołowy typu 1 jest zazwyczaj po raz pierwszy ujawnia się w ciągu pierwszych 10 roku życia, rozwój ziarniniakowe kandydozy (kandydozy skóry i błony śluzowej) i niedoczynności przytarczyc. W przyszłości (często kilkadziesiąt lat) związane przewlekłej niewydolności nadnerczy (choroba Addisona). Kryteriów klinicznych dla diagnozy zespołu jest kombinacja dwóch lub trzech wyżej zaburzeń wydzielania wewnętrznego.
Kandydoza w APG-1 jest uogólnioną i wpływa na błony śluzowe jamy ustnej, narządów rodnych, dróg oddechowych i przewodu pokarmowego, skóry i paznokci, rolki, paznokci. Niedoczynność przytarczyc objawia drgawki mięśni, parestezje skóry, laringospazmom, drgawki, przypomina padaczka. Niewydolność nadnerczy u pacjentów z autoimmunologicznym zespołem wzrasta wieloguczołowe ukrytych i mogą być przedstawione po raz pierwszy na tle kryzysu addisonicheskim stanu naprężenia.
Klasyczna triada wielogruczołowy autoimmunologicznego zespołu jest często związane z hipogonadyzm pierwotny (45%) łysienie (30%) zespół złego wchłaniania (23%) anemia złośliwa (14%) przewlekłe autoimmunologiczne zapalenie wątroby (12%) autoimmunologiczne zapalenie tarczycy (10%), cukrzyca insulinozależna (4%) bielactwo (4%). Rzadziej dystrofia płytki paznokcia, niedorozwój szkliwa, kłębuszkowe zapalenie nerek, astma oskrzelowa, zaćma.
Na tle autoimmunologicznym zespołem wielogruczołowy typu 1 u kobiet obchodzony niedorozwój jajnika (Autoimmunologiczne zapalenie jajnika), przy pierwszorzędowej lub drugorzędowej brak menstruacji. Hipogonadyzm męski przejawia zmniejszenie libido, impotencja, bezpłodność.
Objawy autoimmunologiczne zespół wielogruczołowy typu 2
Manifestacja autoimmunologicznych wielogruczołowy typu zespół 2 kont dla dojrzałego wieku (około 30 lat). Pierwszym objawem endokrynopatią zwykle przewlekłą niewydolność nadnerczy. Inne składniki autoimmunologicznych (cukrzyca typu 1, autoimmunologiczne zapalenie tarczycy), zwykle zgadzają się na 7-10 lat lub więcej.
U pacjentów z autoimmunologicznym zespołem wielogruczołowy typu 2 często rozwija się hipogonadyzm pierwotny, miastenię, bielactwo, zapalenie skóry, łysienie autoimmunologiczne zapalenie żołądka, celiakia, steatorrhea, poliserozita (zapalenie opłucnej, zapalenie osierdzia, wodobrzusze) grasiczak. może wystąpić zespół wielogruczołowy autoimmunologiczna zanik nerwu wzrokowego, guza przysadki, autoimmunologiczna plamica i t. d.
Najczęściej spotykane w wersji praktyce klinicznej autoimmunologicznego zespołu wielogruczołowego typu 2, który łączy główny przewlekłą niewydolność nadnerczy z autoimmunologicznych tarczycy zmian: autoimmunologiczne zapalenie tarczycy, co najmniej - dyfuzję toksyczne wole (Syndrom Schmidta).
Stwardnienie endokrynopatią wzajemnie nasilać siebie i pogorszyć przebieg choroby.
Diagnosttika autoimmunologiczny zespół wielogruczołowy
Kryteria klinicznej diagnostyki laboratoryjnej i potwierdza instrumentalnych pojedynczych składników autoimmunologiczny zespół wielogruczołowy (śluzówkowo kandydoza i niedoczynności przytarczyc w APG hnn hnn-1, autoimmunologiczne zapalenie tarczycy i APG-2 cukrzycy). Gdy APG Typ 1 utrzymywania najbardziej znaczący cząsteczkowego analizy genetycznej w celu identyfikacji charakterystycznych mutacji genu.
Badania laboratoryjne autoimmunologiczny zespół wielogruczołowy polega na określeniu biochemiczne krwi (poziom całkowitego i zjonizowanego wapnia, fosforu, potasu, sodu, bilirubiny, aminotransferaz, fosfatazy alkalicznej, całkowitego białka, mocznika, kreatyniny, glukozy) CBS krwi i wsp. Ważne informacje diagnostyczne dają badania hormonów: TTG, wolnej tyroksyny, hormonu przytarczyc, ACTH, insulina, C-peptyd, kortyzol, renina, aldosteron, somatomedyna C TPO przeciwciała do komórek beta trzustki i GAD testoster to, LH, FSH i inne.
Badania instrumentalne obejmują USG jamy brzusznej, tarczycy, narządów miednicy (u kobiet) i moszny u mężczyzn, echokardiografia, nadnercza CT. Gdzie wskazano konsultacje specjalistów - diabetologa, Gastroenterologii, Hepatologii, Dermatolog, mykolog, neurolog, hematologa, okulisty, ginekologa, endokrynologa, androlog, reumatologa.
Leczenie autoimmunologicznego zespołu wielogruczołowy
Terapia z autoimmunologicznym zespołem wielogruczołowy jest zadaniem złożonym i składa się z leczenia poszczególnych składników. Podstawą patogenetycznego terapii ciągłej hormonalnej terapii zastępczej w niewydolności funkcjonalnej wpływ gruczołów dokrewnych. W przypadku niewydolności nadnerczy powołane glukokortykoidy (deksametazon, hydrokortyzon, prednizolon, triamcynolon), mineralokortykoidów (Doxey, trimetylooctanu deoksykortykosteronu i in.), W niedoczynności tarczycy - L-tyroksyny.
Kiedy kandydozy towarzyszący autoimmunologiczna zespół wielogruczołowy typu 1, stosowane leki przeciwgrzybicze. Przy cukrzycy autoimmunologicznej przeciw wielogruczołowy zespół typu 2 może wymagać leczenia immunosupresyjnego cyklosporyną.
Pacjentom zaleca się stosowanie zwiększonej ilości kwasu askorbinowego i jego sole. Zakazane spożywanie alkoholu, niektóre leki.
Rokowanie autoimmunologicznego zespołu wielogruczołowy
Wczesne wykrycie wielogruczołowy autoimmunologicznego zespołu i przeprowadzenia odpowiedniej terapii zastępczej może kontrolować chorobę. Jednakże, zdolność do pracy na ogół zmniejsza się - pacjentów przypisanych do II-III niepełnosprawności.
U pacjentów z autoimmunologicznym zespołem wielogruczołowy zastrzeżeniem endokrynologa poradni obserwacji i innych specjalistów. W stresujących warunkach, współistniejącymi zakażeniami, fizyczne lub psychiczne pacjentów strain trzeba zwiększyć dawkę hormonów. Potrzeba natychmiastowego leczenia u lekarza jakiegokolwiek pogorszenia stanu zdrowia. Letalny efekt zespołu wielogruczołowego autoimmunologicznych może nastąpić od krtani trzewny kandydozy, ostrej niewydolności nadnerczy.
 Achlorhydria
Achlorhydria Autoimmunologiczne zapalenie wątroby
Autoimmunologiczne zapalenie wątroby Samica pseudohermafrodytyzm
Samica pseudohermafrodytyzm Hiperandrogenizm u kobiet
Hiperandrogenizm u kobiet Niewydolność hormonalna u kobiet
Niewydolność hormonalna u kobiet Hashimoto
Hashimoto Niewydolność nadnerczy
Niewydolność nadnerczy Hashimoto
Hashimoto Panhypopituitarism
Panhypopituitarism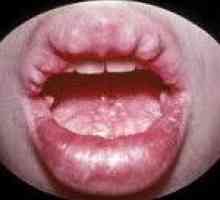 Zespół wielokrotnego gruczolakowatości
Zespół wielokrotnego gruczolakowatości Tarczycy
Tarczycy Autoimmunologiczne zapalenie tarczycy (choroba Hashimoto)
Autoimmunologiczne zapalenie tarczycy (choroba Hashimoto)- Niedoczynność tarczycy
 Choroby autoimmunologiczne
Choroby autoimmunologiczne- Określenie poziomu kortyzolu
- Określenie poziomu dehydroepiandrosteronu
- Określenie poziomu testosteronu
- Test z kortykotropiny
- Test z deksametazonem
 Ciąża z autoimmunologiczną chorobą wątroby (zapalenie wątroby, zapalenie dróg żółciowych, marskość)
Ciąża z autoimmunologiczną chorobą wątroby (zapalenie wątroby, zapalenie dróg żółciowych, marskość)- Aldosteron
 Panhypopituitarism
Panhypopituitarism Hiperandrogenizm u kobiet
Hiperandrogenizm u kobiet Niewydolność hormonalna u kobiet
Niewydolność hormonalna u kobiet Choroby autoimmunologiczne
Choroby autoimmunologiczne Achlorhydria
Achlorhydria Zespół wielokrotnego gruczolakowatości
Zespół wielokrotnego gruczolakowatości Samica pseudohermafrodytyzm
Samica pseudohermafrodytyzm